

Are you a budding secondary instrumental teacher looking to better understand the most common counting system?
Do you wonder if this system could help your elementary students?
There’s a reason this system is one of the most common with instrumentalists around the world. But what is 1 e and a counting?
The 1 e and a counting system uses division and subdivision of syllables to help students and musicians read rhythms in various meters. This system uses syllables and numbers to help students and musicians better perform rhythms, especially complicated ones.
Read on for a breakdown of the system.

Save time with these 60 FREE Music Resources to use in your room right away!
Stop searching the whole internet to find good activities. I’ll help you cut to the chase with my favorite 60 FREE resources.
Table of Contents
1 E And A: The Breakdown
The 1 e and a system depends on the time signature written.
(See the Quick Definitions section towards the end if you come across a word you’re unfamiliar with.)
For those unfamiliar with time signatures, the general rule is this:
- The top number refers to the number of beats in a measure.
- The bottom number refers to the type of rhythm that gets the beat.
So, in 4/4 time, the top number (4) indicates there are four beats in each measure. The bottom number (4) indicates that a quarter note (¼) gets the beat. So 4 quarter notes make up one measure and the quarter note gets the number.
In 3/8, there are 3 beats per measure, and the eighth note gets the beat.
This isn’t a hard and fast rule as 6/8 can be counted in two beats with a triple division or as 6 slow beats. These rule-breakers depend on the tempo of the piece and the conductor’s discretion.
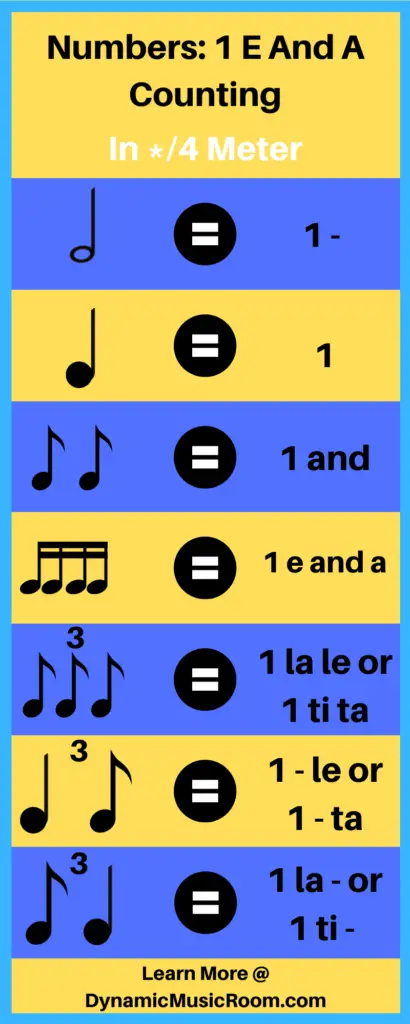
Every beat is given a number to match the top number of the time signature.
Example: 4/4 time. Four quarter notes would be counted “1,2,3,4”.
Division of the beat is counted with “ands”.
Example: 4/4 time. Eight eighth notes would be counted “1 and 2 and 3 and 4.”
Subdivision of the beat is counted with “e” for the first and “a” for the second.
Example: 4/4 time. 16 sixteenth notes would be counted “1 e and a 2 e and a 3 e and a 4 e and a.”
Triplets or division in triple meter are counted with either “1 la le” or “1 trip-let.”
Example: 6/8 time signature. 6 eighth notes would be counted “1 la le 2 la le” or “1 trip-let 2 trip-let.”
Here is a quick reference list for how to count in the 1 e and a system:
- Beat = say the ## of the beat
- Division = and
- Subdivision = ## e and a
- Further divisions don’t merit counting (too fast)
- Triple division = 1 la le or 1 trip-let
- Triplet subdivisions are usually beyond counting
- Rests are left silent and counted internally
All of this may seem like a lot, but it’s easier to see in examples. See the next section for more practical examples.
How Do You Count Rhythms? 4 Examples
In this section, we’ll take the ideas from above and apply it to some practical examples. Use these ideas to check your own understanding of the system.
#1 Basic In Duple

This example covers the basic rhythms in duple time signatures. Notice that notes longer than the beat are just held on the original syllable.
#2 Basic In Triple

This example covers the basic rhythms in triple time signature.
#3 Dotted Rhythms In Duple

Dotted rhythms should be called by the first syllable or number they land on.
#4 Triplets In Duple

Triplets in duple meter are counted just like they would be in triple. Triplet rhythms longer than a beat can be read as “tri-po-let.”
Quick Definitions
In this section, we’ll give some quick definitions for some rhythm terms you may be unfamiliar with. These definitions aren’t intended to fully explain each concept, but just give you a working understanding.
Time Signature (Meter) – This is the fraction-looking number at the beginning of the music. The top number tells how many beats there are in a measure, while the bottom tells you the type of rhythm that gets the beat.
Measure – The music is divided up into block called measures. These measures contain a certain number of beats depending on the time signature.
Beat – The beat is the pulse of music. In regards to rhythm and notation, the bottom number of the time signature tells you which type of rhythm aligns with the pulse.
Division – The next smallest unit the beat is split into. In duple, the beat is split into two, but in triple, the beat is split into three.
Subdivision – The further splitting of the division. Typically, this is into two again, but it could be three.
Another way to think of this is to split the beat into four (in duple) or six (in triple).
Duple (Simple) – This meter is when the beat is naturally split into 2 divisions. Common time signatures include:
- 2/2
- 4/4
- 3/4
Triple (Compound) – This meter is when the beat is naturally split into 3 divisions.
- 6/8
- 9/8
What’s The Origin Of The 1 E And A Rhythm Counting System?
There isn’t a clear origin to this counting system. It’s believed to have evolved from the instrumental director’s counting off “1, 2, 3, 4” to come in.
It also could have been born from the dance world. This group often uses complicated movement depending on the beat and time a song is based in.
These movements would be helped by the breaking down or dividing of beats.
As for the specific “and”s and “e a”s, it’s unclear where this comes from, exactly. It’s theorized that some of these ideas were drawn from the older rhythm counting systems (see below).
Other Rhythm Counting Systems
1 e and a may be the most popular with instrumental programs, but it’s not the only one out there in use. There are many rhythm counting systems available.
Here are a few of the common counting systems you may also want to check out:
- Kodaly Rhythm Syllables (ta ti ti rhythms)
- Froseth Du-de (in use by Music Learning Theory and Conversational Solfege)
- Takadimi
- French Time-Name (a common Orff rhythm syllable)
- TaKeTiNa
Conclusion
I hope you enjoyed learning about the 1 e and a rhythm counting system. This system has the ability to grow with the students into more complicated music and is very popular in instrumental programs.
Looking for songs to use with counting rhythms? Check out these 30 favorite songs eBook to use with your music class.